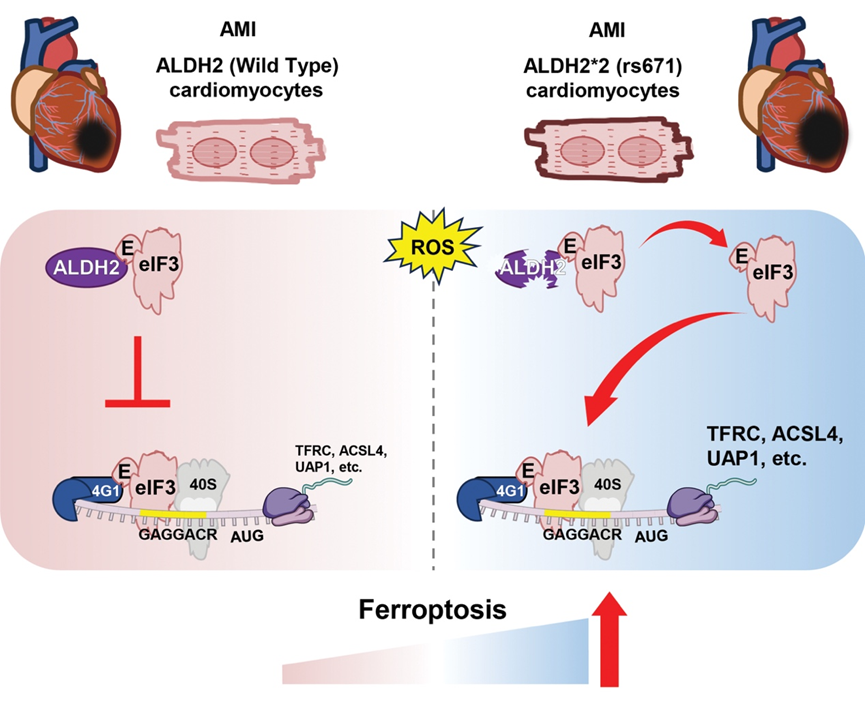
On 20 Oct. 2025, Professor Huiyong YIN’s lab from the Department of Biomedical Sciences published a groundbreaking study published in Circulation entitled ALDH2/eIF3E Interaction Modulates Protein Translation Critical for Cardiomyocyte Ferroptosis in Acute Myocardial Ischemia Injury. This study, for the first time, reveals that ALDH2*2 aggravates acute heart failure post-MI by promoting the selective translation of ferroptosis-related mRNAs (see Figure below). ALDH2 deficiency due to ALDH2*2 mutation disrupts its interaction with the eukaryotic translation initiation factor 3 (eIF3) complex through the eIF3E factor to assemble an eIF3E–eIF4G1–mRNA ternary complex. This remodeling selectively enhances the translation of key ferroptosis regulators, including TFRC and ACSL4, thereby promoting cardiomyocyte ferroptosis and worsening cardiac outcomes.
Ferroptosis, an iron-dependent form of regulated cell death driven by lipid peroxidation, has been implicated in ischemic heart injury. However, the mechanisms underlying ferroptosis in acute myocardial infarction (AMI) remain poorly understood. Acetaldehyde dehydrogenase 2 (ALDH2) detoxifies lipid aldehydes and acetaldehyde from alcohol consumption. A common single nucleotide polymorphism (SNP), ALDH2 rs671 (ALDH2*2) with Glu504Lys replacement, represents one of the most common human SNPs, affecting around 8% global populations with alcohol flush syndrome. In Eastern Asians, around 40% of populations carry this SNP. Previous epidemiological and clinical data have linked ALDH2*2 to an increased risk of MI. However, how ALDH2*2 mechanistically exacerbates cardiac dysfunction after MI remains elusive.
In a Chinese cohort of 177 acute heart failure patients, ALDH2*2 carriers exhibited more severe post-MI cardiac dysfunction. Lipidomic profiling revealed elevated ferroptosis-associated bioactive lipids and decreased levels of antioxidants, such as coenzyme Q10 (CoQ10) and tetrahydrobiopterin (BH4) in plasma. These findings provide clinical metabolomic evidence linking ferroptosis to acute heart failure in ALDH2*2 carriers.
In MI mouse models, ALDH2*2 knock-in mice exhibited aggravated heart failure. Treatment with the ferroptosis inhibitor Fer-1 markedly improved cardiac function and reduced ferroptosis markers. Mechanistically, wild-type ALDH2 binds to the eIF3 complex via eIF3E, thereby preventing eIF3E–eIF4G1 assembly and restricting selective mRNA translation. The ALDH2*2 variant disrupts this interaction, freeing eIF3E to assemble translation complexes that selectively enhance mRNAs harboring the GAGGACR motif (e.g., TFRC, ACSL4, UAP1), leading to ferroptosis. Consistently, in primary cardiomyocytes, ALDH2*2 deficiency similarly promoted eIF3E-dependent selective translation and ferroptosis, further validating this mechanism and highlighting a previously unrecognized non-enzymatic role of ALDH2. Moreover, cardiomyocyte-specific eIF3E knockdown rescued cardiac function and suppressed ferroptosis in ALDH2*2 mice.
In summary, this study uncovers a previously unrecognized role of ALDH2 in regulating ferroptosis via selective mRNA translation control. Targeting ferroptosis represents a promising therapeutic strategy to mitigate post-MI cardiac injury, particularly in ALDH2*2 carriers.
This study was led by former PhD students Drs. Xin Chen and Xiujian Yu at Shanghai Institute of Nutrition and Health (SINH), Chinese Academy of Science, and Dr. Shanshan Zhong, research Associate at BMS. Scientists from SINH, Naval Medical University, Sun Yat-sen University, Fudan University, and Harbin Medical University contributed to this work. This study was financially supported by grants from National Natural Science Foundation of China, Shenzhen Medical Research Fund, RGC, and City University of Hong Kong.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.125.075220
Leave a Reply